笔记本实验
本笔记本利用Amazon Braket实现了 Quantum Molecular Unfolding 和 Molecular Unfolding with Quantum Annealing 。
分子对接
分子对接 (MD) 是药物发现过程的重要步骤,旨在计算一个分子彼此结合时的首选位置和形状。此步骤侧重于计算模拟分子识别过程。它旨在实现蛋白质和配体的优化构象以及蛋白质和配体之间的相对方向,从而使整个系统的自由能最小化。
在这项工作中,蛋白质或口袋被认为是刚性结构,而配体被认为是 灵活的原子集
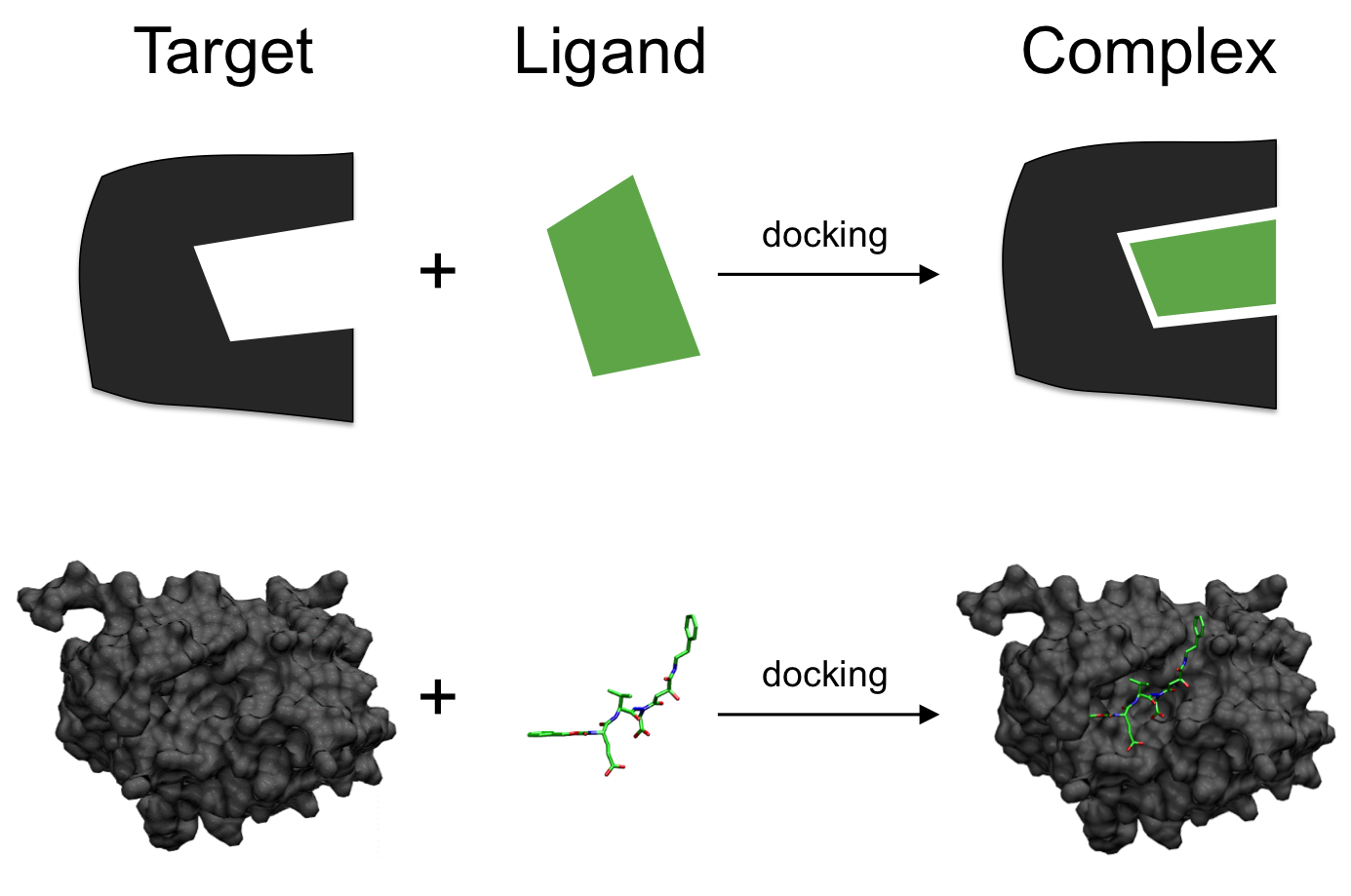
图 6: 分子对接6
正如 Mato 等人发表的 Quantum Molecular Unfolding 中所述,MD 中通常有三个主要阶段:
-
将配体展开成展开的形状,以减少偏差,即分子展开(MU)。包括:
- 可旋转键的识别
- 内部距离最大化
- 消除与工具相关的偏见(例如,SMILES-to-3D)
-
初始安置,包括:
- 配体主要片段分解
- 配体初始姿势识别
- 用刚性旋转平移将配体放入口袋中
-
形状细化,包括:
- 使用可旋转键修改配体形状并匹配蛋白质口袋
- 对接分数最大化
在这里,我们专注于 MD 的第一阶段,即配体扩展,其目的是减少可能影响最终对接质量的形状偏差。在此解决方案中,我们使用 Mato 等人发表的量子退火方法进行分子展开 (MU)。
部署完成后,您可以在堆栈页面选择解决方案的根堆栈,选择输出(Outputs),打开笔记本的链接。请到healthcare-and-life-sciences/a-1-molecular-unfolding-quadratic-unconstrained-binary-optimization/molecular-unfolding-qubo.ipynb查看细节。